Dissolution Testing: How FDA Ensures Generic Drug Quality
 Feb, 9 2026
Feb, 9 2026
The FDA doesn't rely on human trials for every generic drug. Instead, it uses a smart, science-backed tool called dissolution testing to make sure generics work just like the brand-name version. This isn't just paperwork-it's the backbone of how the agency confirms that a $5 pill delivers the same medicine, at the same speed, as its $50 counterpart.
Why Dissolution Testing Matters
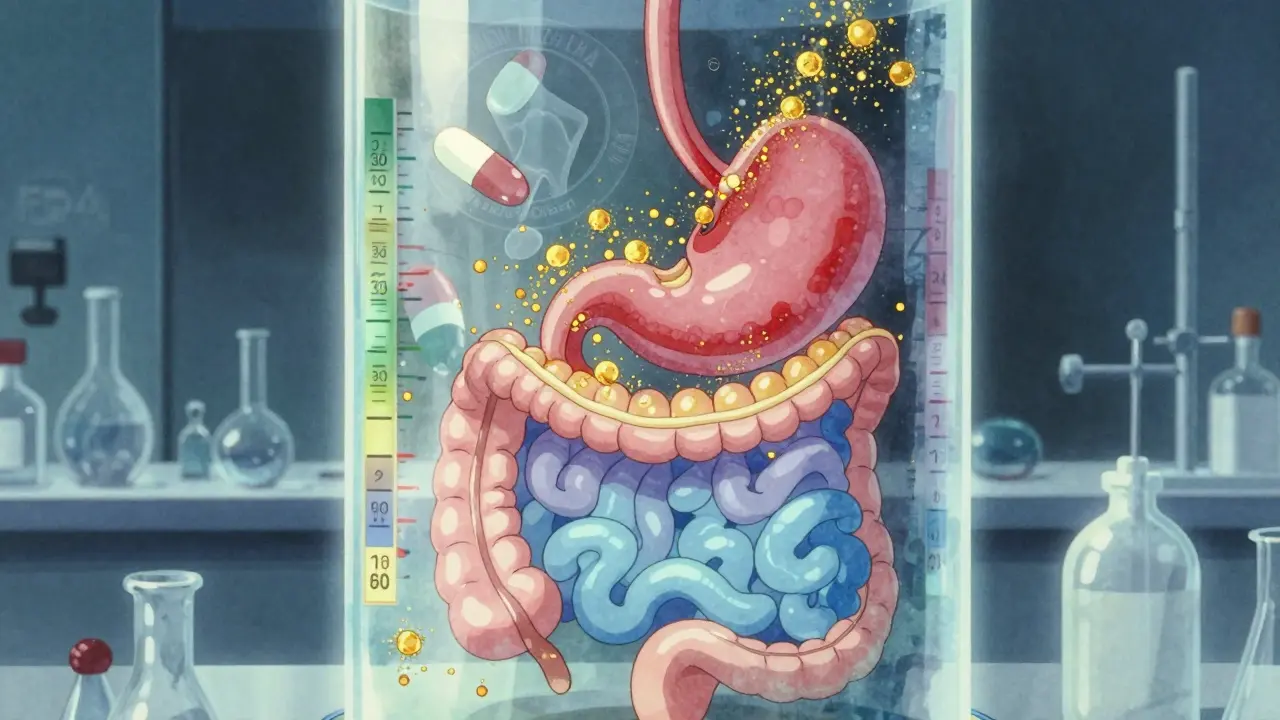
Imagine a pill that looks identical to the brand-name drug but releases its active ingredient too slowly-or too fast. That could mean it doesn't work, or worse, it causes side effects. Dissolution testing measures how quickly and completely a drug dissolves in a lab setting that mimics the human digestive system. The FDA requires this test for nearly all oral solid drugs-tablets, capsules, and suspensions-because if the drug doesn't dissolve properly, your body won't absorb it correctly.
For generic manufacturers, this test replaces expensive and time-consuming human bioequivalence studies. The FDA knows that if two drugs dissolve at the same rate under controlled conditions, they'll likely behave the same inside your body. This saves time, money, and reduces the need to expose volunteers to experimental drugs.
How the FDA Sets the Rules
The FDA doesn't use one-size-fits-all standards. Each drug has its own dissolution profile based on its chemistry, formulation, and intended release pattern. For immediate-release tablets, the standard is often simple: at least 80% of the drug must dissolve within 45 minutes. But for drugs with low solubility, like some cholesterol or blood pressure medications, the test gets more complex. These may require multiple pH levels, different agitation speeds, or even alcohol challenges to simulate what happens if someone takes the pill with a drink.
The agency uses a metric called the f2 similarity factor to compare the dissolution curve of the generic drug to the brand-name version. An f2 score of 50 or higher means the two profiles are statistically similar. That’s the green light for approval. If the score is below 50, the manufacturer must revise the formulation or provide additional data.
For high-solubility drugs (BCS Class I), the FDA allows a biowaiver. That means if the generic dissolves within 30 minutes in 0.1N HCl (a simulated stomach fluid), no human study is needed. This shortcut exists because these drugs are easily absorbed regardless of formulation. It’s a win for patients and manufacturers alike-faster access, lower costs.
What Goes Into a Dissolution Test
It’s not just dropping a pill in water. The FDA requires manufacturers to submit detailed evidence across five areas:
- Solubility of the active ingredient-how well the drug dissolves in different pH environments.
- Test conditions-which apparatus (usually USP Apparatus 1 or 2), rotation speed (often 50-100 rpm), fluid volume (500-900 mL), and pH buffer are used.
- Method validation-proving the test reliably measures the drug’s release, even if conditions change slightly.
- Analytical method accuracy-ensuring the lab equipment can precisely detect how much drug is dissolved at each time point.
- Discriminatory power-the test must be sensitive enough to catch differences between good and bad formulations.
This isn’t optional. The FDA expects 50 to 100 pages of documentation in each ANDA application just for dissolution testing. Manufacturers spend months-sometimes over a year-perfecting these methods before even submitting their application.

Modified-Release Drugs: A Bigger Challenge
Extended-release pills are trickier. They’re designed to release medication slowly over hours. If they release too fast-say, after someone drinks alcohol-they can cause overdose. That’s why the FDA requires alcohol challenge tests. A generic version of an extended-release opioid, for example, must be tested in fluids with up to 40% ethanol to ensure it doesn’t “dose-dump.”
These products also need testing at multiple pH levels: stomach acid (pH 1.2), upper intestine (pH 4.5), and small intestine (pH 6.8). Each condition simulates a different part of the digestive tract. A drug that dissolves perfectly in water might fail in the acidic environment of the stomach. The FDA’s Dissolution Methods Database includes specific protocols for over 2,800 drug products, helping manufacturers avoid guesswork.
The Role of the FDA’s Database
The FDA maintains a public database of recommended dissolution methods. As of late 2023, it includes approved test conditions for 2,847 drug products across 1,245 unique formulations. This isn’t just a reference-it’s a lifeline for generic manufacturers. Instead of starting from scratch, they can follow a proven method, saving months of development time.
When a manufacturer uses a method from the database, they still need to validate it for their specific product. But they don’t have to prove the entire method from scratch. This reduces regulatory burden without sacrificing quality.

What Happens When Dissolution Doesn’t Match?
Even if two drugs dissolve differently, the FDA doesn’t automatically reject the generic. Sometimes, the brand-name product itself has a flawed dissolution profile. In those cases, the FDA may approve the generic with different specifications. This happened with several generic versions of metformin and other common drugs. The agency focuses on therapeutic equivalence, not identical lab curves.
Manufacturers must also prove their dissolution profile doesn’t change if they switch suppliers, change an excipient, or move production lines. The SUPAC-IR guidelines require comparative testing at multiple time points to ensure consistency. A change that alters the release rate by even 5% can trigger a full FDA review.
Where the System Is Headed
The FDA is moving toward more physiologically relevant testing. That means using fluids that better mimic real human digestion-like enzymes, bile salts, and food effects. Early data shows this improves prediction accuracy, especially for poorly soluble drugs.
There’s also growing interest in expanding biowaivers. In 2022, the FDA began exploring whether BCS Class III drugs (high solubility, low permeability) could qualify for dissolution-based approval. If approved, this could cut development time for dozens of new generics.
By 2025, experts estimate 35% of generic approvals will use standardized dissolution methods-up from 25% in 2020. But the FDA’s stance remains firm: dissolution must be product-specific. There are no shortcuts around scientific rigor.
Why This System Works
Dissolution testing isn’t perfect-but it’s the best tool we have. It’s fast, repeatable, and cost-effective. It lets the FDA approve hundreds of generic drugs each year without putting patients at risk. And because it’s grounded in real science, not assumptions, it’s trusted across the global regulatory community.
When you pick up a generic pill, you’re not just saving money. You’re benefiting from a regulatory system that uses chemistry, physics, and precise measurement to guarantee quality. The FDA doesn’t guess. It measures. And that’s what keeps millions of people safe every day.
What is dissolution testing in generic drugs?
Dissolution testing is a laboratory procedure that measures how quickly a drug releases its active ingredient under controlled conditions that simulate the human digestive system. For generic drugs, it’s used to prove the product releases the drug at the same rate and extent as the brand-name version, ensuring therapeutic equivalence without requiring human trials.
How does the FDA use dissolution testing to approve generics?
The FDA compares the dissolution profile of a generic drug to the reference listed drug (RLD) using metrics like the f2 similarity factor. If the profiles are statistically similar (f2 ≥ 50), the generic is considered bioequivalent. For certain drugs, especially high-solubility ones, a successful dissolution test can eliminate the need for human bioequivalence studies entirely.
Are all generic drugs required to undergo dissolution testing?
Yes, all oral solid dosage forms-tablets, capsules, suspensions-must undergo dissolution testing. The FDA exempts only drugs already in solution, like oral liquids or topical formulations, because their release isn’t controlled by dissolution. Even for these, other quality controls apply.
What is the f2 similarity factor?
The f2 similarity factor is a statistical tool used by the FDA to compare dissolution profiles between two products. It ranges from 0 to 100, with 50 or higher indicating the profiles are similar enough to be considered bioequivalent. A score below 50 usually means the generic formulation needs adjustment.
Can a generic drug be approved even if its dissolution profile differs from the brand?
Yes. If the brand-name drug itself has a poor or inconsistent dissolution profile, the FDA may approve a generic with different specifications, as long as it demonstrates therapeutic equivalence through clinical data or other evidence. The goal is patient safety and effectiveness-not identical lab curves.
What role does the FDA’s Dissolution Methods Database play?
The database provides manufacturers with pre-approved dissolution test conditions for over 2,800 drug products. It saves time and reduces uncertainty by offering validated methods, so companies don’t have to develop them from scratch. While manufacturers still need to validate the method for their specific product, the database significantly streamlines the approval process.
Marie Fontaine
February 9, 2026 AT 22:32Love how the FDA uses science instead of guesswork! Seriously, who else would’ve thought to measure pill dissolution like a chemistry lab? This is why generics work & why I don’t stress about switching brands. $5 vs $50? Same results. 🙌
Brandon Osborne
February 11, 2026 AT 00:05Oh wow, so the FDA just lets drug companies test pills in water and calls it a day? No human trials? That’s insane. What if the pill dissolves in the lab but explodes in your stomach? You think they’re testing this on real people or just robots in a basement somewhere? This is how people die. I’ve seen it. And now they’re saving money by skipping the real science? Pathetic.
MANI V
February 11, 2026 AT 09:38Wow, you people actually believe this? Dissolution testing? Please. The FDA is a puppet of Big Pharma. They approve generics based on lab numbers while real patients get sick because the pills don’t dissolve the same way in real life. And you call this science? It’s corporate theater. They don’t care about you. They care about profit. This isn’t regulation-it’s a license to kill.
Random Guy
February 12, 2026 AT 10:02So you’re telling me a $5 pill dissolves in 45 minutes and somehow works the same as a $50 one? Bro. I took a generic for my anxiety and felt like I was being slowly drained by a zombie. Coincidence? Nah. I’m not buying this fairy tale anymore.
Ken Cooper
February 13, 2026 AT 09:48ok so i just read this whole thing and wow. i had no idea they tested pills in different ph levels like stomach acid and stuff. like imagine if your pill just dissolved in water but in your body it’s all acidic?? that’d be bad. and the f2 factor? sounds like a nerdy math thing but it’s actually super smart. also the alcohol challenge?? that’s wild. i never thought about someone drinking a beer with their pill. yikes. glad someone’s checking this stuff. also the database with 2800+ methods?? that’s insane. like, who even makes that? scientists with too much time??
Tasha Lake
February 13, 2026 AT 21:03The dissolution profile is the critical quality attribute for BCS Class I and III drugs. The f2 similarity factor is validated under ICH Q1(R2) guidelines, and the use of USP Apparatus II at 75 rpm with pH 1.2, 4.5, and 6.8 buffers ensures robust discrimination. The SUPAC-IR criteria further mandate that any change in excipient sourcing must demonstrate <5% deviation in dissolution kinetics at t=15, 30, and 45 min. This is regulatory science at its finest.
Sam Dickison
February 14, 2026 AT 15:14Appreciate the breakdown. I’ve worked with generics in pharmacy and honestly? The dissolution testing is the unsung hero. Most people think it’s just ‘same pill, cheaper’-but nope. The formulation tweaks, the pH buffers, the alcohol challenges? It’s all hyper-specific. One time a manufacturer switched from HPMC to PVP and the release profile went sideways. Took 6 months to fix. This system isn’t perfect, but it’s way more rigorous than most realize.
Brett Pouser
February 15, 2026 AT 09:28As someone from a country where generics are just… sold without testing, this blew my mind. In my hometown, people buy pills from roadside vendors. No dissolution, no f2, no nothing. I used to think the US was over-regulating. Now I see-this is what safety looks like. Thank you, FDA. Seriously.
John McDonald
February 15, 2026 AT 12:07Love how this isn’t just about saving money-it’s about making sure people aren’t getting sick because a pill didn’t dissolve right. I’ve had friends who got weird side effects from generics and blamed the brand. Turns out it was a bad batch. This system catches that. Good job, science.
Ryan Vargas
February 16, 2026 AT 01:21Let me ask you this: if dissolution testing is so foolproof, why do we still have recalls? Why do lawsuits pile up over generic drug failures? Because the FDA’s database is built on assumptions, not reality. The lab environment is sterile. The human body is chaos. Enzymes fluctuate. Gut flora changes. Food interacts. The f2 score is a statistical illusion masking a systemic risk. They call it ‘science’-but it’s a mathematical fantasy built on 1970s assumptions. And now they’re expanding biowaivers? You’re not saving time-you’re gambling with lives. This isn’t regulation. It’s a Ponzi scheme dressed in lab coats.
Andrew Jackson
February 16, 2026 AT 22:12It is a matter of profound national concern that the United States of America, a beacon of scientific integrity, has permitted the erosion of its pharmaceutical oversight through the mechanistic substitution of human bioequivalence studies with laboratory dissolution metrics. This represents a dangerous capitulation to global cost-cutting agendas. While the f2 similarity factor may appear statistically valid, it fails to account for the nuanced, dynamic physiology of the American patient. We do not test on rats. We do not test on beakers. We test on human beings. And yet, the FDA has abdicated its sacred duty. This is not innovation. This is surrender. And history will not forgive us.